Structural protein stability within the cell as a new Achilles heel to tackle antibiotic resistance
A MAECI-MinCyt joint project between CERM (Florence, Italy) and IBR (Rosario, Argentina)
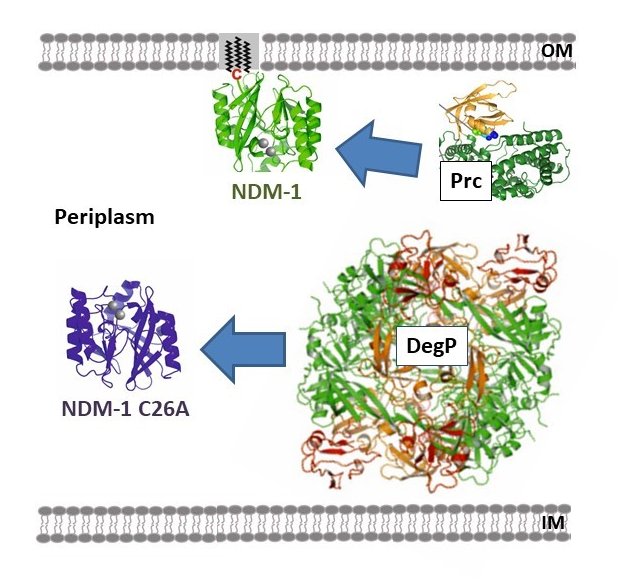 Antibiotic resistance is nowadays one of the most serious public health threats. This problem is rapidly accelerating due to the worldwide spread of antibiotic-resistance genes, leaving few options for clinical treatment. Within this context Carbapenems are last resort b-lactam antibiotics. However, carbapenems are inactivated by zinc-dependent metallo-b-lactamases (MBLs), such as Bacillus cereus II (BcII) and New Delhi metallo-b-lactamase (NDM-1), present in multi-resistant bacterial pathogens. The host innate immune elicits a response to bacterial infection that involves sequestering of essential metal ions, such as Zn(II), from the pathogens. We have recently shown that this response triggers the degradation of MBLs within bacteria, rendering them susceptible to antibiotics. Different MBLs display distinct stabilities upon metal restriction: while soluble NDM-1 is readily degraded, soluble BcII and membrane anchored NDM-1 are refractory to this process. In this project we seek to understand the intracellular metabolic fate of two paradigmatic lactamases (BcII and NDM-1) under conditions of variable Zn(II) availability. To this aim, we will exploit novel in-cell NMR techniques and microbiology tools to unravel the folding and Zn(II) uptake mechanisms of MBLs within the bacterial cell. In addition, we will characterize how metal ion deprivation results in enzyme degradation and how membrane anchoring modulates protein stability.
Antibiotic resistance is nowadays one of the most serious public health threats. This problem is rapidly accelerating due to the worldwide spread of antibiotic-resistance genes, leaving few options for clinical treatment. Within this context Carbapenems are last resort b-lactam antibiotics. However, carbapenems are inactivated by zinc-dependent metallo-b-lactamases (MBLs), such as Bacillus cereus II (BcII) and New Delhi metallo-b-lactamase (NDM-1), present in multi-resistant bacterial pathogens. The host innate immune elicits a response to bacterial infection that involves sequestering of essential metal ions, such as Zn(II), from the pathogens. We have recently shown that this response triggers the degradation of MBLs within bacteria, rendering them susceptible to antibiotics. Different MBLs display distinct stabilities upon metal restriction: while soluble NDM-1 is readily degraded, soluble BcII and membrane anchored NDM-1 are refractory to this process. In this project we seek to understand the intracellular metabolic fate of two paradigmatic lactamases (BcII and NDM-1) under conditions of variable Zn(II) availability. To this aim, we will exploit novel in-cell NMR techniques and microbiology tools to unravel the folding and Zn(II) uptake mechanisms of MBLs within the bacterial cell. In addition, we will characterize how metal ion deprivation results in enzyme degradation and how membrane anchoring modulates protein stability.
The project is funded by ![]()
WebNMR4 is a web application to manage NMR Instruments, its planning, members, projects, etc. It's possible to do statistics and so on.
You can create users with different rule that give them different privileges.
You can preview the planning with different cells color depending on the special character used.
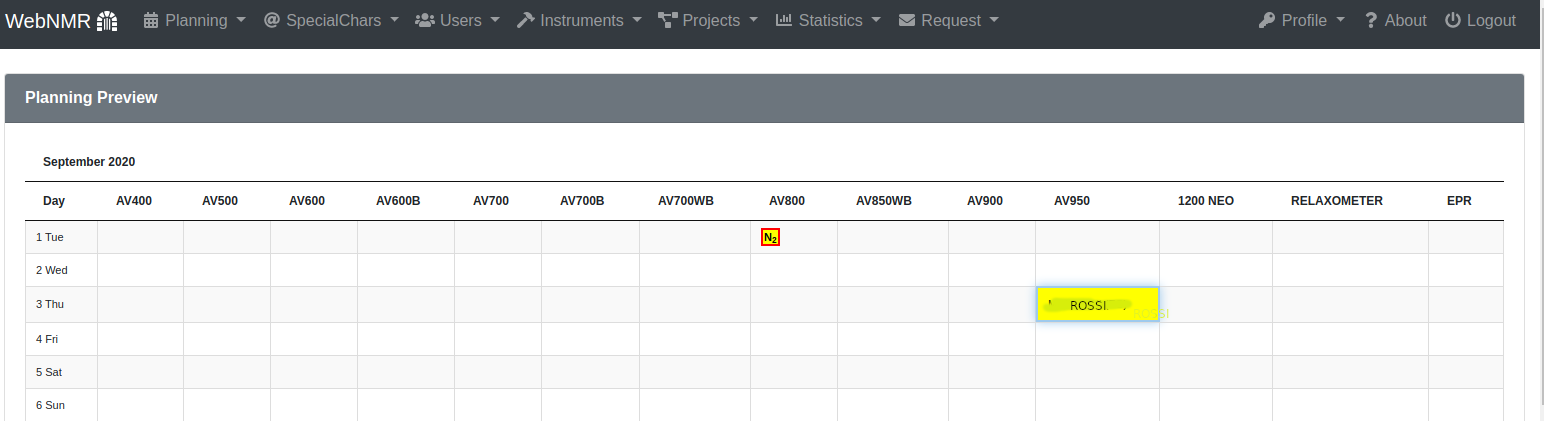
You can associate a special code to a color/user pair. Inserting the code in a cell you'll see the colored cell in the preview.

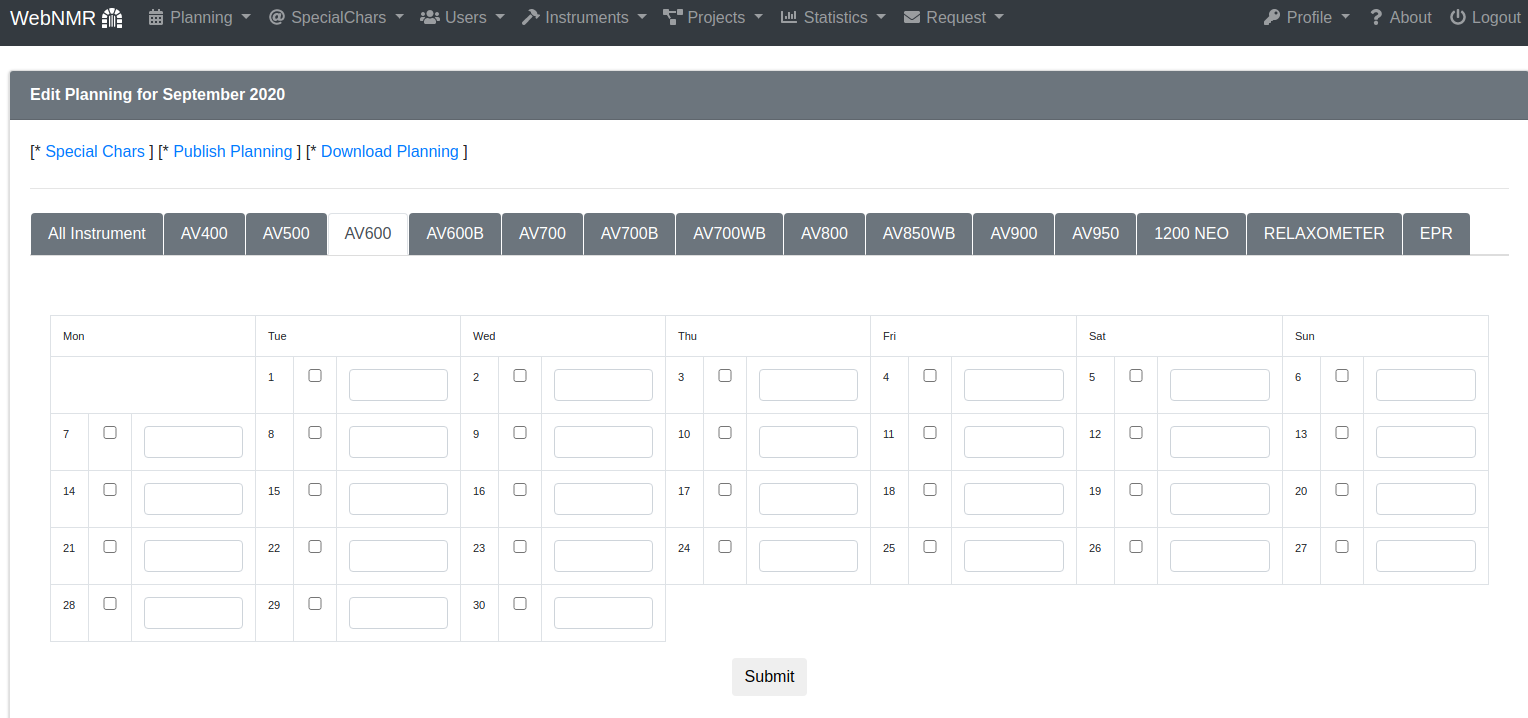
It's possible to manage planning for a single instrument..
..or for all instruments. You have to select the cell and write. When you press enter the data are saved.
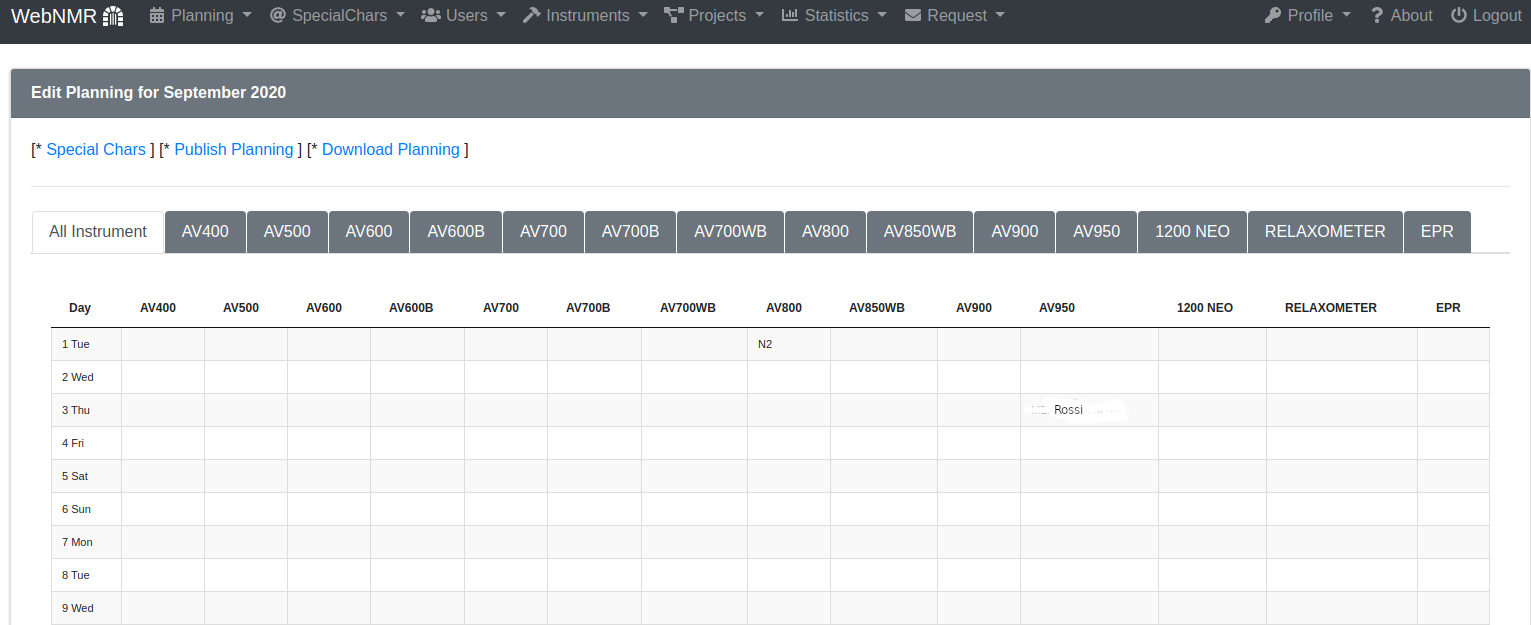
You can edit and preview more month and you can select the period that a normal user can see in the first page or selecting Published from the Planning menu.
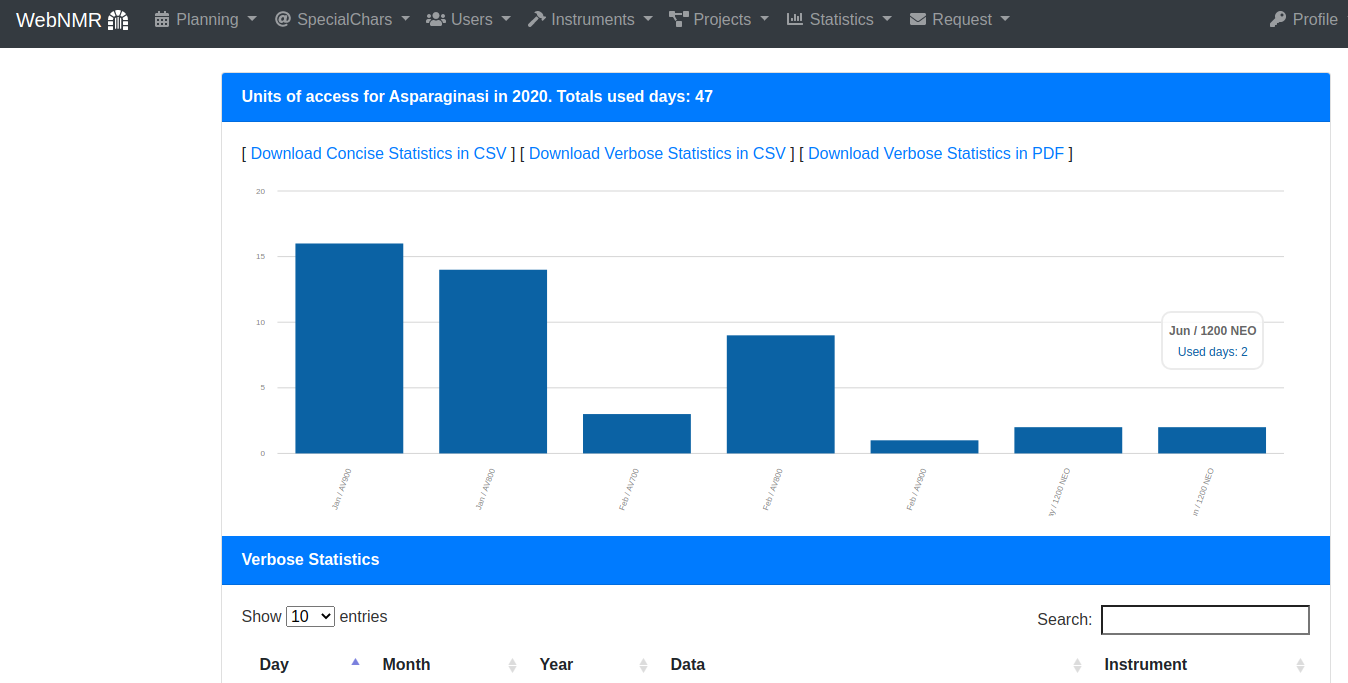
It's possible to do statistics.
New modules of the program CYANA (developed at the ETH, Zurich) allowing the user to exploit pseudocontact shifts, residual dipolar couplings and cross-correlation rates in paramagnetic systems as structural restraints. These new modules have been developed at the Florence RI.
A patch has been created, to upgrade from Cyana to the new version:
From CYANA to Paramagnetic-CYANA
REFERENCE
Balayssac, Bertini, Luchinat, Parigi, Piccioli, J. Am. Chem. Soc. (2006) 128, 15042-15043.
PARArestraints for Xplor-NIH is the complete set of modules which have been developed in order to use paramagnetism-based NMR restraints in protein structure calculations with the program Xplor-NIH. Paramagnetism-based restraints are paramagnetic relaxation enhancements, pseudocontact shifts, residual dipolar couplings due to metal and overall magnetic anisotropy, and cross-correlation between Curie relaxation and nuclear-nuclear dipolar relaxation.
User Manual:
Tutorial (.doc)
Tutorial (.pdf)
A patch has been created, to upgrade Xplor-NIH version 2.9.2 with the new modules:
You can also download the source files:
Here you can find two sample calculations (files updated on 02/03/2004):
When using PARArestraints for Xplor-NIH, please cite:
Banci L, Bertini I, Cavallaro G, Giachetti A, Luchinat C, Parigi G, 2004, "Paramagnetism-based restraints for Xplor-NIH", Journal of Biomolecular NMR 28: 249-61.
A new software tool for FBDD by NMR
The benchmarking study of software tools conducted in the first part of the project revealed that the majority of the programs for automated analysis of spectra described in the literature were not of immediate availability to users for FBDD studies; some require the local installation of a program, upon request to the authors, whereas others are embedded in complex, sometimes commercial, suites. Therefore, CIRMMP decided to develop at home a new dedicated software tool (PICASSO)1 that compares two spectra, one of the free protein and one of the small molecule-protein mixture, based on the corresponding peak lists. The performance of the tool in terms of correct identification of the protein-binding regions has been evaluated on different protein targets (matrix metalloproteinase 12, human carbonic anhydrase II, human transthyretin, human musashi, human MDM2, human HUR) using experimental data from interaction studies with a set of selected ligands (19) already published or available. For the systems investigated, our tool achieved between 79% and 100% correct assignments, properly identifying the protein regions involved in the interaction.
Fragment screening NMR pipeline web server: an on-line accessible workflow for FBDD by NMR
To further reduce the manual involvement and to maximize the output of NMR fragment screening campaign, the software tool PICASSO has been integrated in an automated workflow. The service allows for the automatic identification of binding ligands to protein targets as well as the evaluation of per residue chemical shift perturbations between the free protein spectrum and the ligand-bound protein spectrum. In particular, the assignment of the spectrum of the free protein and the raw data of the spectrum of the ligand-bound protein are needed. The service takes as input NMR spectra in the Bruker format and automatically processes them using the NMRPipe software. The 2D 1H–15N correlation spectra are analyzed and assigned using Picasso software, and the chemical shift perturbation data are provided as output in CSV format.
Fragment screening NMR pipeline web service is now available at the link: https://nmrfrascan.cerm.unifi.it/frontend